





11.1 Introduction
Acetylcholine, the first neurotransmitter discovered, was originally described as "vagus stuff" by Otto Loewi because of its ability to mimic the electrical stimulation of the vagus nerve. It is now known to be a neurotransmitter at all autonomic ganglia, at many autonomically innervated organs, at the neuromuscular junction, and at many synapses in the CNS.
In this chapter we will discuss the acetylcholine’s anatomy, cell biology, physiological effects, role in behavior, and clinical applications.
Figure 11.1 |
||
11.2 Acetylcholine in the Autonomic Nervous System
In the autonomic nervous system, acetylcholine (ACh) is the neurotransmitter in the preganglionic sympathetic and parasympathetic neurons. These are shown in Figure 11.2 as the red ACh in the ganglion. ACh is also the neurotransmitter at the adrenal medulla and serves as the neurotransmitter at all the parasympathetic innervated organs. ACh is also the neurotransmitter at the sweat glands, and at the piloerector muscle of the sympathetic ANS (Labeled in blue in Figure 11.2).
Figure 11.2 |
11.3 ACh in the Peripheral Nervous System
In the peripheral nervous system, ACh is the neurotransmitter at the neuromuscular junction between the motor nerve and skeletal muscle.
Figure 11.3 |
11.4 ACh in the Central Nervous System
In the central nervous system, ACh is found primarily in interneurons, shown in Figure 11.3 as orange and green cell clusters. A few important long-axon cholinergic pathways have also been identified. Noteworthy is the cholinergic projection from the nucleus basalis of Meynert (in the basal forebrain) to the forebrain neocortex and associated limbic structures, represented by the black pathway in Figure 11.3. Degeneration of this pathway is one of the pathologies associated with Alzheimer's disease. There is also a projection from the medial septal and diagonal band region to limbic structures (blue). Most subcortical areas are innervated by neurons from the ponto-mesencephalic region (purple in Figure 11.3).
11.5 Introduction to the Cell Biology of the Cholinergic Synapse
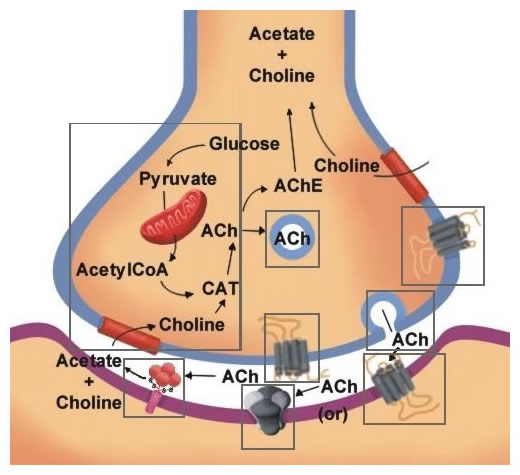
Figure 11.4 is a summary of the biological mechanisms involved in the synthesis, storage secretion, receptor interaction and termination of acetylcholine. Click on the region of the cell describing these processes to learn more about each one.
|
Figure 11.4 |
Click on the blocks marked (above) to see details, or choose from the list below.
- Synthesis of Acetylcholine
- Storage of Acetylcholine
- Release of Acetylcholine
- Acetylcholine Receptors
- Termination of Acetylcholine Action
11.6 Synthesis of ACh
Figure 11.5 |
Choline acetyltransferase (CAT): As shown in Figure 11.5, ACh is synthesized by a single step reaction catalyzed by the biosynthetic enzyme choline acetyltransferase. As is the case for all nerve terminal proteins, CAT is produced in the cholinergic cell body and transported down the axon to the nerve endings. Both CAT and ACh may be found throughout the neuron, but their highest concentration is in axon terminals. The presence of CAT is the "marker" that a neuron is cholinergic, only cholinergic neurons contain CAT.
The rate-limiting steps in ACh synthesis are the availability of choline and acetyl-CoA. During increased neuronal activity the availability of acetyl-CoA from the mitochondria is upregulated as is the uptake of choline into the nerve ending from the synaptic cleft. Ca2+ appears to be involved in both of these regulatory mechanisms. As will be described later, the inactivation of ACh is converted by metabolism to choline and acetic acid. Consequently much of the choline used for ACh synthesis comes from the recycling of choline from metabolized ACh. Another source is the breakdown of the phospholipid, phosphatidylcholine. One of the strategies to increase ACh neurotransmission is the administration of choline in the diet. However, this has not been effective, probably because the administration of choline does not increase the availability of choline in the CNS.
11.7 Storage of ACh
The majority of the ACh in nerve endings is contained in clear (as viewed in the electron microscope) 100 um vesicles. A small amount is also free in the cytosol. Vesicle-bound ACh is not accessible to degradation by acetylcholinesterase (see below).
The uptake of ACh into storage vesicle occurs through an energy-dependent pump that acidifies the vesicle. The acidified vesicle then uses a vesicular ACh transporter (VAChT) to exchange protons for ACh molecules. No useful pharmacological agents are available to modify cholinergic function through interaction with the storage of ACh.
Interestingly, the gene for VAChT is contained on the first intron of the choline acetyltransferase gene. This proximity implies the two important cholinergic proteins are probably regulated coordinately.
Figure 11.6 |
11.8 Release of ACh
The release of ACh occurs through Ca2+ stimulated docking, fusion, and fission of the vesicle with the nerve terminal membrane, as discussed previously.
You will recall that the miniature endplate potentials and the quantal release in response to action potentials at the neuromuscular junction are due to the release of packets of ACh from individual storage vesicles (Chapter 5). Many toxins are known that interfere with these processes and are effective in preventing ACh secretion. The examples in Figure 11.6 shows botulinum toxin inhibition and black widow spider venom (BWSV) stimulation of ACh release.
Figure 11.7 |
11.9 ACh Receptors
There are two broad classes of cholinergic receptors: nicotinic and muscarinic. This classification is based on two chemical agents that mimic the effects of ACh at the receptor site nicotine and muscarine.
Table I summarizes some of the properties of nicotinic and muscarinic receptors.
Table I |
|
| Nicotinic | Muscarinic |
| Bind nicotine | Bind muscarine |
| Blocked by curare (tubocurarine) | Blocked by atropine |
| Linked to ionic channels | Linked to 2nd messenger systems through G proteins (see below) |
| Response is brief and fast | Response is slow and prolonged |
| Located at neuromuscular junctions, autonomic ganglia, and to a small extent in the CNS | Found on myocardial muscle, certain smooth muscle, and in discrete CNS regions |
| Mediate excitation in target cells | Mediate inhibition and excitation in target cells |
| Postsynaptic | Both pre- and postsynaptic |
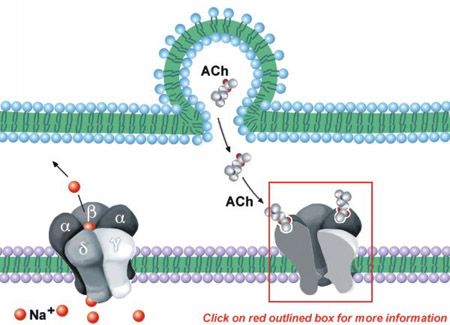
11.10 The Nicotinic Receptor is an Ion Channel
|
Figure 11.8 |
As indicated in Table I, nicotinic receptors are located at the NMJ, autonomic ganglia and sparsely in the CNS.
The NMJ nicotinic ACh receptor consists of five polypeptide subunits: two α subunits and one each of β, δ, and γ (see Figure 11.8). A funnel-shaped internal ion channel is surrounded by the five subunits. The binding surface of the receptor appears to be primarily on the α subunits, near the outer surface of the molecule. The subunits contain recognition sites for agonists, reversible antagonists, and α-toxins (cobra α-toxin and α-bungarotoxin).
Whereas the NMJ nicotinic receptor is composed of four different species of subunit (2 α, β, γ, δ), the neuronal nicotinic receptor also is composed of only two subunit types (2 α and 3 β).
11.11 The Muscarinic Receptor is Coupled to G-Proteins
Figure 11.9 |
Muscarinic receptors, classified as G protein coupled receptors (GPCR), are located at parasympathetic autonomically innervated visceral organs, on the sweat glands and piloerector muscles and both post-synaptically and pre-synaptically in the CNS (see Table I). The muscarinic receptor is composed of a single polypeptide. Seven regions of the polypeptide are made up of 20-25 amino acids arranged in an α helix. Because each of these regions of the protein is markedly hydrophobic, they span the cell membrane seven times as depicted in Figure 11.9. The fifth internal loop and the carboxyl-terminal tail of the polypeptide receptor are believed to be the site of the interaction of the muscarinic receptor with G proteins (see right). The site of agonist binding is a circular pocket formed by the upper portions of the seven membrane-spanning regions.
ACh has excitatory actions at the neuromuscular junction, at autonomic ganglion, at certain glandular tissues and in the CNS. It has inhibitory actions at certain smooth muscles and at cardiac muscle.
Figure 11.10 |
The biochemical responses to stimulation of muscarinic receptor involve the receptor occupancy causing an altered conformation of an associated GTP-binding protein (G protein). G protein is made up of the three subunits α, β and γ. In response to the altered conformation of the muscarinic receptor, the a subunit of the G protein releases bound guanosine diphosphate (GDP) and simultaneously binds guanosine triphosphate (GTP). The binding of GTP "activates" the G protein, allowing dissociation of the α subunit from the trimeric complex and for it to interact with effector systems to mediate specific responses. An inherent GTPase catalytic activity of the G protein hydrolyzes the GTP back to GDP. This hydrolysis terminates the action of the G protein. The rate of hydrolysis of the GTP thus dictates the length of time the G protein remains activated.
The responses mediated by muscarinic receptors through G proteins include:
Figure 11.11 |
Inhibition of Adenylate Cyclase: The muscarinic receptor, through interaction with an inhibitory GTP-binding protein, acts to inhibit adenylyl cyclase. Reduced cAMP production leads to reduced activation of cAMP-dependent protein kinase, reduced heart rate, and contraction strength.
Figure 11.12a |
Figure 11.12b |
Stimulation of Phospholipase C: The muscarinic receptor activates phosphoinositide-specific phospholipase C (PLCβ) through interaction with a GTP-binding protein. As shown in Figure 11.12a, the hydrolysis of phosphatidylinositol bisphosphate yields two second messengers; inositol trisphosphate (IP3) and diacylglycerol (DAG). The DAG activates protein kinase C (not shown). Cellular responses are influenced by PKC's phosphorylation of target proteins. As shown in Figure 11.12b, the IP3 diffuses to the smooth endoplasmic reticulum (ER) where it interacts with IP3 receptors to increase Ca2+ release from the intracellular storage site.
Figure 11.13 |
Activation of K+ Channels: In response to muscarinic cholinergic receptor stimulation, a GTP binding protein also can interact directly with K+ channels to increase K+ conductance, (Figure 11.13). This conductance increase increases the resting membrane potential in myocardial and other cell membranes leading to inhibition.
11.12 Termination of ACh Action
Figure 11.14 |
ACh binds only briefly to the pre- or postsynaptic receptors. Following dissociation from the receptor, the ACh is rapidly hydrolyzed by the enzyme acetylcholinesterase (AChE) as shown in Figure 11.14. This enzyme has a very high catalysis rate, one of the highest known in biology. AChE is synthesized in the neuronal cell body and distributed throughout the neuron by axoplasmic transport. AChE exists as alternatively spliced isoforms that vary in their subunit composition. The variation at the NMJ is a heteromeric protein composed of four subunits coupled to a collagen tail that anchors the multi-subunit enzyme to the cell membrane of the postsynaptic cell (Figure 11.14). This four-subunit form is held together by sulfhydryl bonds and the tail anchors the enzyme in the extracellular matrix at the NMJ. Other isoforms are homomeric and freely soluble in the cytoplasm of the presynaptic cell. AChE, unlike ChAT, is found in non-cholinergic neurons as well. In addition, other cholinesterases exist throughout the body, which are also able to metabolize acetylcholine. These are termed pseudocholinesterases.
Drugs that inhibit ACh breakdown are effective in altering cholinergic neurotransmission. In fact, the irreversible inhibition of AChE by isopropylfluoroesters are so toxic that they can be incompatible with life—inhibiting the muscles for respiration. This inhibition is produced because ACh molecules accumulate in the synaptic space, keep the receptors occupied, and cause paralysis. Two notable examples are insecticides and the gases used in biological warfare. The mechanism of action of these irreversible inhibitors of AChE is that they carbamylate the AChE, rendering it inactive. The carbamylation inactivates both the acetyl and choline binding domains. A recently developed antidote to these inhibitors cleaves the nerve gas so that it will dissociate from the AChE.
In contrast to the irreversible inhibitors, the reversible AChE inhibitors are effective in transiently increasing the ACh level and are effective in diseases and conditions where an increased ACh level is desired. The clinically important compound, eserine (physostigmine), reversibly inhibits AChE.
11.13 Physiology
Nicotinic receptor activation causes the opening of the channel formed by the receptor. This increases the Na+ movement into the target cell, leading to depolarization and generation of the action potential. This rapidly developing change, termed a fast EPSP, is illustrated in Figures 4.3, and 6.2.
Figure 11.15 |
Muscarinic receptor activation of postsynaptic cells can be either excitatory or inhibitory and is always slow in onset and long in duration (Table I). Figure 11.16 and Figure 11.17 illustrate an excitatory and an inhibitory postsynaptic potential in the sympathetic ganglion. As described earlier, G protein activation underlies all actions of the muscarinic receptors, thus accounting for their slow onset.
Figure 11.16 |
Figure 11.17 |
|
Slow EPSP and IPSP form the sympathetic ganglion of the rat. |
||
11.14 Behavior
The rapid nature of the synaptic transmission mediated by the nicotinic receptor is consistent with its role at the NMJ and in the ganglion of the ANS. Little is known about the role of the nicotinic receptor role in CNS behavior. Clearly, nicotine stimulation is related in some manner to reinforcement, as indicated by the prevalence of nicotine addiction among humans.
Muscarinic receptors, in contrast, are important mediators of behavior in the CNS. One example is their role in modulating motor control circuits in the basal ganglia. A second example is their participation in learning and memory. The latter is inferred from two types of observations: 1) muscarinic antagonists are amnesic agents, and 2) deterioration of the cholinergic innervation of the neocortex is associated with memory loss in Alzheimer’s disease.
11.15 Clinical
Alzheimer's disease: A disease in which a marked deterioration occurs in the CNS, the hallmark of which is a progressive dementia. One of the characteristics of this disease is a marked decrease in ACh concentrations in the cerebral cortex and caudate nucleus.
Myasthenia gravis: A disease of the neuromuscular junction in which the receptors for ACh are destroyed through the actions of the patient's own antibodies.
Cholinergic Pharmacology: Numerous drugs are used clinically to interact with the cholinergic systems. Table II summarizes the major uses for cholinergic drugs.
11.16 Cholinergic Pharmacological Agents
| Table II Cholinergic Pharmacological Agents |
||
| Drug | Action | Clinical Use |
| Atropine (and other anticholinergics) | Blocks muscarinic receptors | Relaxes muscle in the eye causing the pupil to dilate. Used when the eye is inflamed and during eye examinations. |
| Slows the activity of the stomach and intestinal track and reduces acid secretion. Therefore, used for stomach cramps, diarrhea, diverticulitis, pancreatitis, bed wetting, motion sickness. | ||
| There has been some indication of this drug for Parkinson’s disease. | ||
| Scopolamine | Blocks CNS muscarinic receptors | Used topically to prevent dizziness, nausea and other aspects of motion sickness. |
| Amantadine (Symmetrel) | Blocks muscarinic receptors | Antidyskinetics used to treat Parkinson’s disease and the dyskinesia associated with antipsychotic drugs |
| Bethanechol | Mimics ACh | Used to treat urinary retention, and stimulate movement of intestinal tract. |
| Tacrine (Cognex) | Blocks ACh breakdown | Treat Alzheimer’s disease |
| Eserine or physostigmine | Blocks ACh breakdown | Reduces pressure in the eye and is used to treat glaucoma |
| Used to diagnose and treat myasthenia gravis | ||
Test Your Knowledge
Which of the following is effective in increasing the level of acetylcholine in the synapse or neuromuscular junction? (NOTE: There is more than one correct answer.)
Which of the following is effective in increasing the level of acetylcholine in the synapse or neuromuscular junction? (NOTE: There is more than one correct answer.)
Which of the following is effective in increasing the level of acetylcholine in the synapse or neuromuscular junction? (NOTE: There is more than one correct answer.)
Which of the following is effective in increasing the level of acetylcholine in the synapse or neuromuscular junction? (NOTE: There is more than one correct answer.)
Which of the following is effective in increasing the level of acetylcholine in the synapse or neuromuscular junction? (NOTE: There is more than one correct answer.)
Which of the following is effective in increasing the level of acetylcholine in the synapse or neuromuscular junction? (NOTE: There is more than one correct answer.)
|
