The improvement in overall health care has brought about an increase in the life span of the population and created the problem of dealing with the health care needs and diseases that affect the elderly. This situation has unmasked Alzheimer's disease, a formidable disease that will be discussed in detail later in this chapter.
10.1 Aging Is a Normal Biological Process
Maximal and average life expectancy -- Aging is a normal biological process that is programmed into the genetics of all species. Thus aging, per se, is not caused by disease. Each animal species has a maximal life expectancy that is hypothesized to be unchangeable. Figure 10.1 is a survival curve for humans illustrating that the maximal life expectancy is approximately 90 years of age. In contrast to maximal life expectancy, the average life expectancy of individuals of a species can be dramatically influenced by environmental influences, especially disease. Thus, Figure 10.1 also shows that the average life expectancy has changed markedly. The figure shows that the average life expectancy, represented by “fifty percent survivors” has increased from 47 years in 1900 to 70+ years in 1989.
Figure 10.1 |
10.2 Biological Changes in the Absence of Disease in Aging Humans
Below are listed some of the biological changes that occur in aging humans in the absence of disease.
Reductions in the following sensory functions:
- temperature regulation
- high frequency hearing, taste, smell, feel, touch & pain sensitivity
- transparency of the optical light path
Examples of major changes in proteins with age:
- collagen become less elastic
- muscle protein is reduced (muscular strength and autonomic tone decrease)
- levels of brain neurotransmitter synthesizing enzyme decrease
General changes in aging:
- function of the immunological system deceases
- neuronal conduction velocity decreases
- O2 consumption decreases
- ATP synthesis rate decreases
- reduced thermal regulation
- reduced cardiovascular regulatory function
Behavioral changes in aging (These usually are not marked):
- reduced short-term memory
- bouts of forgetfulness
- transient confusion
- errors in judgment
- reduced learning rate
- decreased ability to respond to stress
CNS parameters that decrease in aging:
- brain weight and volume - 15%
- gray matter content; for example 50% of the motor neurons in the anterior horn may disappear by age 60 years
- Nissl substance reflecting decreased mRNA in neurons
- dendritic arborization and spine number
- dendrite diameter
- chromatin staining
- melanin pigment (retina, locus ceruleus and substantia nigra)
- synaptic vesicle number in some neurons
CNS parameters that increase in aging:
- ventricular volume
- astroglia number and size
- myelin sheath thickness
- lipofuscin (formerly termed aging pigment)
- neuritic plaques and neurofibrillary tangles
10.3 Alzheimer's Disease (AD)
Alzheimer’s disease (AD) has become the most common neurodegenerative disorder, currently affecting nearly twenty five million worldwide. AD, which begins with mild memory deficits and modest neuronal death, is followed by progressive extensive neuronal death, eventually leading to severe dementia. In addition, AD is frequently accompanied by other neurological and personality impairments including slow movements, hampered motor coordination, general confusion and personality change. AD patients generally live for about 9 years after initial clinical diagnosis. Thus treatment is necessary for extensive time periods. This not only places a great burden on caregivers, but the economy as well, with an annual cost yearly of $160 billion worldwide. AD is estimated to affect over four million people in the USA (10% of the population over 65 and 47% of those over 85). The pathological features of Alzheimer’s disease, described below, are associated with profound degeneration of neurons and synapses in a few regions of the CNS, including the temporal, parietal, and frontal cortices, all of which are associated with learning and memory processes. The two most characteristic pathological inclusions found in the brains of AD patients are extracellular deposits of β-amyloid peptides (Aβ) that lead to senile plaque formation and intracellular neurofibrillary tangles of hyperphosphorylated tau. However, increasing evidence has suggested that inflammation may play a critical role in AD pathogenesis as well.
10.4 Diagnosis of Alzheimer's Disease
In this chapter, the changes that occur with AD and the way to clinically diagnose the disease are summarized as a means to describe the disease and to illustrate how the physician must clinically evaluate the possibility of AD. According to the Psychiatric Manual three progressive stages or categories of AD diagnosis are defined. These include: 1) Suspected AD, 2) Probable AD, and 3) Definite AD. Definite AD can only be diagnosed after pathological brain specimens are examined by either biopsy or autopsy. The characteristics used to diagnose AD are listed below.
Suspected AD. The following are changes that would lead to the diagnosis of Suspected AD (note that many of the behavioral changes overlap with those occurring in normal aging:)
- loss of mental capacity
- loss of recent memory
- forgetfulness
- transient periods of confusion
- restlessness
- lethargic periods
- errors in judgment
- reduced capacity to learn
Probable AD. Once a patient is determined to meet the criteria for diagnosis of suspected AD; further tests are conducted to establish the likelihood of AD. If the patient satisfies the criteria below, the diagnosis of Probable AD is made.
- clear dementia, (termed senile dementia)-defined as being afflicted by or manifesting unsoundness of mind or an inability to control one's rational processes.
- onset between ages of 40 and 90
- deficits in two or more cognitive spheres
- progressive deterioration of memory and other cognitive functions
- no disturbances of consciousness
- no other illness that could account for progressive cognitive deficits (for example, not cerebral vascular disease)
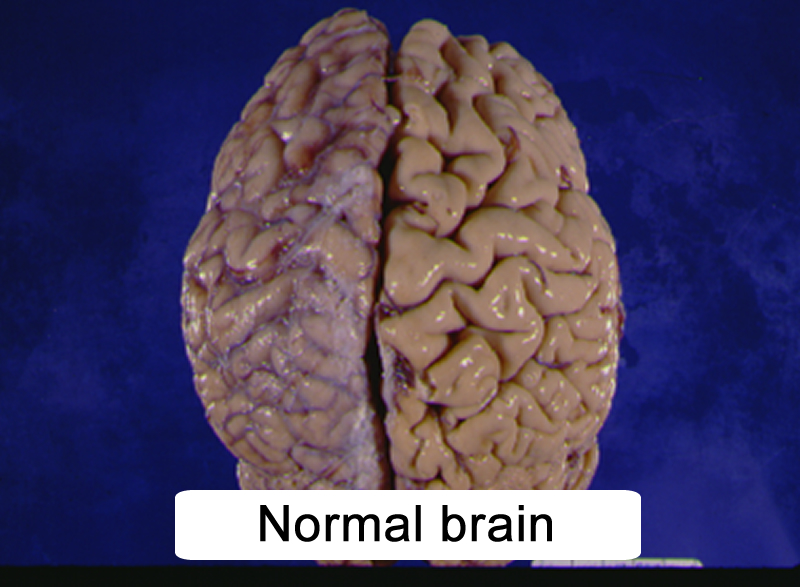 |
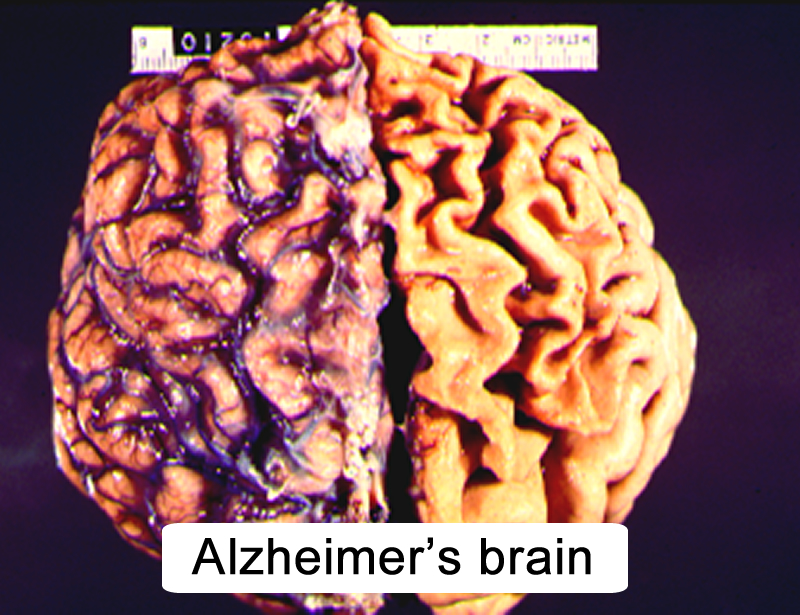 |
Figure 10.2 |
|
Definite AD. To make the diagnosis of Definite AD, morphological pathological inclusions must be present in the patient’s CNS. The nature of the pathological changes that occur in AD and the use of some of these features in the diagnosis of AS are described below.
- atrophied cerebral hemispheres (Figure 10.2)
- dilated ventricles
- Neurofibrillary tangles. These are non-membrane bound masses of fibers in the cell bodies of neurons, shown in Figure 10.3. Neurofibrillary tangles contain many tightly adherent pairs of helical filaments. These are termed paired helical filaments (PHF). Neurofibrillary tangles also contain the same pathological protein, β-amyloid, found in neurite plaques (see below).
Figure 10.3
AD brain showing neurofibrillary tangles and kinky neurites. - Paired helical filaments. Because neurons bearing PHF appear intact and functional, PHF are believed to precede the death of a neuron. Because antibodies to cytoskeleton proteins, such as tau and tubulin, cross-react with PHF it is believed that PHF are composed of modified cytoskeletal proteins. One hypothesis, discussed below, is that PHF are hyper-phosphorylated tau protein. You will recall that tau protein is a microtubule associated protein (MAP)
- Neurite plaques are defined as spherical clusters of dystrophic neurites surrounding a core of β amyloid and are found predominantly in gray matter. (Neurite plaques are also called senile plaques or neuritic plaques). These 10 - 100 µm diameter structures are believed to be remnants of degenerated axon terminals. Because neurite plaques are much more common in the aged Alzheimer's brain than in the normal aged brain, they are considered a specific marker for Alzheimer's disease. A number of neurite plaques are shown in in Figure 10.4.
Figure 10.4
Lower (left) and higher power (right) photomicrographs of senile plaques from an AD brain. - Loss of dendritic branches and spines. As occurs in normal aging, a characteristic feature of AD is a decrease in the number and branches of cortical dendrites and spines. This predicts a decreased synaptic innervation of cortical neurons. In AD there is a large decrease in dendritic branches and spines.
10.5 Diagnosis of Definite AD
With the above pathological features in mind, the physician examines the CNS biopsy or on autopsy to determine the presence of these histopathologic features. Following the examination of the histological sections, the age related criteria in Table I are used to establish the diagnosis of Definite AD. The areas where plaques and tangle are most abundant are usually the temporal- and neo-cortex. The histopathologic analysis requires multiple CNS samples for microscopic examination. Because these structures also occur in normal aging, the diagnosis of AD depends on the number of pathological features based on the patient’s age; with older age, more inclusions are required before AD is indicated. Other regions examined are the amygdala, hippocampus, basal ganglia, substantia nigra, cerebellum, and spinal cord.
| Table I Morphological Observations Necessary to Confirm Alzheimer's Disease at Various Ages |
|
| Age (years) |
Number of Plaques & Tangles (in one sq. mm of neocortical tissue) |
| <50 | must exceed 2-5/field |
| 50-65 | at least 8/field |
| 66-75 | at least 10/field |
| >75 | more than 15/field |
10.6 Classification of Alzheimer's Disease
AD, in few cases, has been found to be associated with specific families. Although this happens rather infrequently, it has led to the classification of AD as either familial or sporadic.
Sporadic AD refers to disease that has no clear-cut genetic basis. It is the majority of AD seen in the general population. However, a genetic predisposition may exist for even this disease, as will be discussed at the end of this chapter.
Familian AD (FAD) refers to an inherited disease that is associated with families. Several examples of AD mapped to a specific gene exist. Based on the typical age of onset, familial AD has been sub-classified as early onset and late onset. Both of these will also be presented in more detail at the end of this chapter.
10.7 Observations Concerning the Causes (Etiology) of Alzheimer's Disease
Historically, several types of observations have been made concerning the cause of AD. Five of these include: 1) abnormally low levels of neurotransmitters, 2) accumulation of a pathological protein, β amyloid, 3) hyperphosphorylation of a microtubule associated protein, tau, 4) products of the presenilin genes and 5) alterations in a cholesterol shuttling protein (ApoE4). Several of these have been analyzed in genetic studies.
- Neurotransmitters. The patterns of biochemical loss in Alzheimer's disease reflect specific involvement of discrete neuronal populations rather than an overall general deterioration. Some of the neurotransmitters that deteriorate include:
- acetylcholine
- norepinephrine
- serotonin
- somatostatin
- acetylcholine
ACH Hypothesis. Among these, the neurotransmitter to receive the greatest attention has been acetylcholine. Morphological and biochemical evidence has implicated a specific deterioration of the cholinergic system that connects the midbrain nucleus basalis of Meynert to the cerebral cortex. The location of the cholinergic neurons is shown in Figure 10.5.
Figure 10.5 |
|
- Beta Amyloid / abnormal protein. An important approach to the study of AD is to analyze the abnormal structures found in the AD brain. One of the principle proteins present is β amyloid, named for its β pleated confirmation. This protein was purified, sequenced and an antibody to a synthetic peptide mimicking the sequence produced. The antibody reacts to cerebral β amyloid and not to other protein in plaques and tangles showing that β amyloid is a constituent of AD neuropathological structures. Isolation of the cDNA encoding β amyloid revealed a number of important properties of this protein as described below.
Figure 10.6 |
β amyloid is a 4,000-4,500 MW protein that is part of a larger 79,000 MW protein. The larger protein is called amyloid precursor protein (APP). Analysis of the hydrophobicity plots predicts that APP spans the cell membrane and extends into the extracellular space (Fig. 11.6).
A number of observations of the biology of amyloid has lead to the hypothesis that amyloid is the toxic substance that initiates the pathology of AD. This hypothesis, called the amyloid hypothesis, is based on morphological, biochemical, and molecular biological evidence that implicates β amyloid as a seminal, causative agent in Alzheimer's disease. The role of β amyloid in Alzheimer’s disease is based on the following observations:
- Amyloid is found in both plaques and tangles and is present in a number of other dementias such as Down's syndrome. Thus, it is hypothesized to be an essential factor in the cause of degeneration.
- Antibodies to β amyloid show pre-amyloid deposits in both Down's syndrome and AD brains. This implies that focal amyloid deposition may be the early event in plaque formation for both these diseases. A mutation in the gene for APP is located on chromosome 21. This gene has been identified in several cases of Familial Alzheimer's disease (FAD). Also, recall that Down’s syndrome patients have three copies of chromosome 21.
Based on the above observations, it is hypothesized that an abnormal proteolytic processing of APP occurs in AD. This abnormal processing is believed to generate a toxic 40-kDa peptide referred to as β amyloid or Aβ (Figure 10.7). Recent research has shown that under normal circumstances a proteolytic enzyme, α secretase, cuts APP to produce a normal biological messenger molecule (Figure 10.7a). However, it appears that in AD, the APP is abnormally proteolyzed by two other secretases (Figure 10.7b) to produce the abnormal protein, β amyloid (Aβ). These two secretases, termed β and γ secretase, have been cloned to determine their biology and structural properties. The goal is to generate drugs that can block the secretases and the initiation or progression of AD. One or the other of these secretases may be the product of the senilin genes of familial AD. Also a recent hypothesis, based on the observation that zinc or copper facilitates the formation of insoluble Aβ, is that drugs that chelate these metals will be a good treatment for AD. Clinical trails are underway examining this hypothesis. Recent (Nature Cell Biology 11, 143-153, 2008) research has shown that there may be an abnormal clearing of Aβ that is responsible for its accumulation and toxicity. This provides another strategy for dealing with AD, enhancing the clearance of Aβ.
How Aβ is toxic to cells is not known. One possibility is that it stimulates increased calcium influx, which can lead to cell death through the hyper-phosphorylation of cytostructural proteins, such as tau (see below). Also, recent evidence indicates that Aβ can stimulate the formation of free radicals that may kill neurons. A single methionine in the Aβ molecule appears to be capable of augmenting free radical formation and neuronal death in neuronal cell cultures. High levels of antioxidants such as vitamin E protect neurons from this adverse effect.
There is also evidence that Aβ may have normal functions in the brain. It has been shown to be: 1) a specific ligand for a number of different receptors and other molecules, 2) transported by complex trafficking pathways, 3) modulated in response to a variety of environmental stressors, and 4) able to induce pro-inflammatory activities. Recent evidence indicates that Aβ may be an antimicrobial peptide that functions normally as a part of an the innate immune system for the brain. How these biological functions relate to the possible role of Aβ in AD is not known.
Figure 10.7a |
Figure 10.7b |
- Tau protein hypothesis - Molecular research over the past ten years suggests that abnormal processing of tau (hyper-phosphorylation) may also be an important aspect of AD pathology. (Figure 10.8) Tau is a microtubule associate protein (MAP) composed of a family of six proteins in mammals. These proteins associate with microtubules in the axons of neurons to regulate microtubule growth, assembly and cross-linking. In normal CNS, MAPs, other than tau, regulate microtubule function in dendrites and the cell soma. However in the degenerating neurons in AD patients, 1) tau is found throughout the neuron, not just in the axon. Further 2) tau is also abnormal in that it is phosphorylated much more extensively in AD than in normal brain. Finally, 3) the tau within degenerating neurons is often associated with PHF. Thus the hyper-phosphorylation of tau may be responsible for the altered subcellular distribution of tau and eventual cell death. These observations point to the abnormal phosphorylation and distribution of tau as an important aspect of the pathology of AD. Importantly, the demonstration that tau is distributed differently in degenerating neurons in Alzheimer’s disease than in normal aging has been interpreted to show that Alzheimer’s disease is not merely the end stage of normal aging. One unifying hypothesis is that tau’s hyperphosphorylation may be induced by the β amyloid.
Figure 10.8 |
|
- Presenilins - Mutations in two additional genes, presenilin-1 (PS-1), located on chromosome 14 and presenilin-2 (PS-2), located on chromosome, have been determined to be a cause of specific forms of FAD. PS-2 is a close homologue of PS1. To date 45 different PS-1 mutations and two PS-2 mutations have been identified in FAD and these mutations account for up 50 % of early-onset FAD. A suggested mechanism of the mutated presenilins is that they facilitate the alternative proteolysis of APP by γ secretase to form Aβ. This conclusion is supported by evidence that mice transfected with only APP developed plaques late in life, whereas mice with both APP and PS-1 mutations show plaques at a much younger age. This suggests that PS-1 ratchets up the pace of the AD disease process.
- ApoE4 hypothesis. ApoE proteins facilitate the shuttling of cholesterol into cells. Researchers at Duke University observed that a specific variant, ApoE4, is elevated in the CSF of Alzheimer’s patients. This observation triggered biochemical and genetic studies of ApoE4 and its role in Alzheimer’s disease. As a consequence, the results of several genetic linkage studies and ApoE4 binding studies linked the ApoE cholesterol shuttling protein to Alzheimer’s disease. These results are:
- The apoE4 gene is positively correlated with several classes of Alzheimer's.
- Of the three variants of ApoE, the ApoE4 variant predominates in familial Alzheimer’s, whereas the ApoE3 form is most common in the general population.
- A similar genetic skew appears in “sporadic” Alzheimer’s.
- The presence of ApoE4 predisposes one to Alzheimer’s. Individuals with two copies of this allele are most likely to contract Alzheimer’s, and to develop it earlier as shown in Figure 10.9.
Figure 10.9 |
10.8 Summary of the Genetics of Alzheimer’s disease
The current information concerning the causes of AFD indicates that it is a genetically heterogeneous disorder. Although most cases of Alzheimer's disease do not exhibit familial inheritance, certain genes appear to act as risk factors for AD. Currently, the best known genetic risk factor for AD is the inheritance of the ε4 allele of the apolipoprotein E on chromosome 19. This gene is implicated in up to 50% of late-onset sporadic Alzheimer's cases. However, geneticists agree that numerous other genes probably also act as risk factors or have protective effects that influence the development of late onset AD. Over 400 genes have been tested for association with late-onset sporadic AD. There is also considerable evidence that Aβ and APP on chromosome 21 play an important role in the etiology of AD. The evidence for genetic basis in FAD and a role for Aβ in sporadic AD include the following: 1) Individuals with Down’s syndrome have three copies of chromosome 21 and consequently make large amount of APP. 2) Down’s syndrome patients exhibit brain pathology virtually identical to AD and suffer dementia. 2) A direct linkage has been made between chromosome 21 and a subset of FAD. 3) In addition a direct linkage has been made between mutations in the genes for presenilins and the facilitation of the abnormal processing of APP to produce Aβ. All of these observations support a role for amaloid in AD pathology.
10.9 Role of Risk Factors and the Environment
Several clear risk factors exist for AD. These include being female, family history of neurodegenerative diseases, less education and less intellectually demanding jobs. How these factors fit into the etiology of AD is not known.
| Table II | ||||
| Type | ||||
| I. Early-onset Familial | II. Early-onset Familial I | III. Early-onset Familial | IV. Late-onset Familian and Sporadic | |
| Chromosome | 21 | 14 | 1 | 19 |
| Gene | APP | PS-1 | PS-2 | ApoE4 |
| Genetic Effect | Direct Linkage | Direct Linkage | Direct Linkage | Risk Factor |
| Typical Age of Onset (years) | 50's | 40's | 50's | Allele-dependent (see Figure 10.5) |
| Incidence Among All Alzheimer's Disease Cases | <20 known families | <2% | Volga-German kindreds | >95% |
- Question 1
- A
- B
- C
- D
The following is evidence that Alzheimer’s disease is not merely exaggerated normal biological aging. Only Alzheimer’s disease brains have:
A. neuritic plaques
B. neurofibrillary tangles
C. hypo-phosphorylated tau
D. tau distributed throughout the neurons
The following is evidence that Alzheimer’s disease is not merely exaggerated normal biological aging. Only Alzheimer’s disease brains have:
A. neuritic plaques This answer is INCORRECT.
Both normal brain and Alzheimer’s brain have neuritic plaques that increase in number with age. In AD the number of neuritic plaques is much higher.
B. neurofibrillary tangles
C. hypo-phosphorylated tau
D. tau distributed throughout the neurons
The following is evidence that Alzheimer’s disease is not merely exaggerated normal biological aging. Only Alzheimer’s disease brains have:
A. neuritic plaques
B. neurofibrillary tangles This answer is INCORRECT.
Both normal brain and Alzheimer’s brain have neurofibrillary tangles that increase in number with age. In AD the number of neurofibrillary tangles is much higher
C. hypo-phosphorylated tau
D. tau distributed throughout the neurons
The following is evidence that Alzheimer’s disease is not merely exaggerated normal biological aging. Only Alzheimer’s disease brains have:
A. neuritic plaques
B. neurofibrillary tangles
C. hypo-phosphorylated tau This answer is INCORRECT.
The neurons in the brains of AD do not have hypophosphorylated tau. It is hyper-phosphorylated.
D. tau distributed throughout the neurons
The following is evidence that Alzheimer’s disease is not merely exaggerated normal biological aging. Only Alzheimer’s disease brains have:
A. neuritic plaques
B. neurofibrillary tangles
C. hypo-phosphorylated tau
D. tau distributed throughout the neurons This answer is CORRECT!
The MAP tau is found throughout neurons in AD, whereas in normal brains this MAP is found only in the axons and NOT in the dendrites.
- Question 2
- A
- B
- C
- D
“Sporadic” Alzheimer’s disease has
A. primarily a genetic cause
B. primarily a environmental cause
C. primarily an infectious cause
D. a combination of genetic, environmental, and infectious causes
“Sporadic” Alzheimer’s disease has
A. primarily a genetic cause This answer is INCORRECT.
Although genetics is believed to be an important determinant of AD, it is thought to be only a portion of the basis for the development of AD. Environmental influences also contribute.
B. primarily a environmental cause
C. primarily an infectious cause
D. a combination of genetic, environmental, and infectious causes
“Sporadic” Alzheimer’s disease has
A. primarily a genetic cause
B. primarily a environmental cause This answer is INCORRECT.
Although environmental influences are believed to be an important determinant of AD, it is thought to be only a portion of the basis for the development of AD. Genetic predisposition is also an important influence.
C. primarily an infectious cause
D. a combination of genetic, environmental, and infectious causes
“Sporadic” Alzheimer’s disease has
A. primarily a genetic cause
B. primarily a environmental cause
C. primarily an infectious cause This answer is INCORRECT.
Although there is evidence that infections and inflammatory reactions are a part of the basis for the development of AD, both environmental influences and genetic predisposition are believed to be important.
D. a combination of genetic, environmental, and infectious causes
“Sporadic” Alzheimer’s disease has
A. primarily a genetic cause
B. primarily a environmental cause
C. primarily an infectious cause
D. a combination of genetic, environmental, and infectious causes This answer is CORRECT!
AD is believed to be caused by the combination of environmental factors, infectious agents and inflammation that come together with the person’s genetic predisposition to produce the disease.
- Question 3
- A
- B
- C
- D
- E
The diagnosis “Definite Alzheimer’s Disease” is established by:
A. autopsy of the patient’s brain
B. behavioral analysis
C. PET analysis
D. CAT analysis
E. MRI analysis
The diagnosis “Definite Alzheimer’s Disease” is established by:
A. autopsy of the patient’s brain This answer is CORRECT!
The psychiatric manual (DSM-IV, Diagnostic and Statistical Manual of Mental Disorders) requires that brain tissue be examined for senile plaques and neuritic tangles to establish "definite Alzheimer's disease".
B. behavioral analysis
C. PET analysis
D. CAT analysis
E. MRI analysis
The diagnosis “Definite Alzheimer’s Disease” is established by:
A. autopsy of the patient’s brain
B. behavioral analysis This answer is INCORRECT.
The psychiatric manual (DSM-IV, Diagnostic and Statistical Manual of Mental Disorders) requires that brain tissue be examined for senile plaques and neuritic tangles to establish "definite Alzheimer's disease".
C. PET analysis
D. CAT analysis
E. MRI analysis
The diagnosis “Definite Alzheimer’s Disease” is established by:
A. autopsy of the patient’s brain
B. behavioral analysis
C. PET analysis This answer is INCORRECT.
The psychiatric manual (DSM-IV, Diagnostic and Statistical Manual of Mental Disorders) requires that brain tissue be examined for senile plaques and neuritic tangles to establish "definite Alzheimer's disease".
D. CAT analysis
E. MRI analysis
The diagnosis “Definite Alzheimer’s Disease” is established by:
A. autopsy of the patient’s brain
B. behavioral analysis
C. PET analysis
D. CAT analysis This answer is INCORRECT.
The psychiatric manual (DSM-IV, Diagnostic and Statistical Manual of Mental Disorders) requires that brain tissue be examined for senile plaques and neuritic tangles to establish "definite Alzheimer's disease".
E. MRI analysis
The diagnosis “Definite Alzheimer’s Disease” is established by:
A. autopsy of the patient’s brain
B. behavioral analysis
C. PET analysis
D. CAT analysis
E. MRI analysis This answer is INCORRECT.
The psychiatric manual (DSM-IV, Diagnostic and Statistical Manual of Mental Disorders) requires that brain tissue be examined for senile plaques and neuritic tangles to establish "definite Alzheimer's disease".
- Question 4
- A
- B
- C
- D
- E
Which of the following is not implicated via direct linkage to “Familial" Alzheimer’s Disease?
A. ApoE4
B. APP
C. Presenilin 1 (PS-1)
D. Presenilin 2 (PS-2)
E. All of the above are implicated by direct linkage.
Which of the following is not implicated via direct linkage to “Familial" Alzheimer’s Disease?
A. ApoE4 This answer is CORRECT!
ApoE4 is a risk factor for Familial Alzheimer’s disease, not a direct link.
B. APP
C. Presenilin 1 (PS-1)
D. Presenilin 2 (PS-2)
E. All of the above are implicated by direct linkage.
Which of the following is not implicated via direct linkage to “Familial" Alzheimer’s Disease?
A. ApoE4
B. APP This answer is INCORRECT.
APP has been shown to have a direct link to Familial Alzheimer's disease.
C. Presenilin 1 (PS-1)
D. Presenilin 2 (PS-2)
E. All of the above are implicated by direct linkage.
Which of the following is not implicated via direct linkage to “Familial" Alzheimer’s Disease?
A. ApoE4
B. APP
C. Presenilin 1 (PS-1) This answer is INCORRECT.
Presenilin 1 has been shown to have a direct link to Alzheimer's Familial disease.
D. Presenilin 2 (PS-2)
E. All of the above are implicated by direct linkage.
Which of the following is not implicated via direct linkage to “Familial" Alzheimer’s Disease?
A. ApoE4
B. APP
C. Presenilin 1 (PS-1)
D. Presenilin 2 (PS-2) This answer is INCORRECT.
Presenilin 2 has been shown to have a direct link to Alzheimer's Familial disease.
E. All of the above are implicated by direct linkage.
Which of the following is not implicated via direct linkage to “Familial" Alzheimer’s Disease?
A. ApoE4
B. APP
C. Presenilin 1 (PS-1)
D. Presenilin 2 (PS-2)
E. All of the above are implicated by direct linkage. This answer is INCORRECT.
APP, Presenilin 1, and Presenilin 2 have been shown to have a direct link to Familial Alzheimer’s disease.
